

文献解析

近日,上海市第一人民医院呼吸与危重症医学科包爱华/张旻团队在《Free Radical Biology and Medicine》上发表重要研究成果。
该研究揭示了慢性氧化应激通过抑制ATF3表达,进而破坏MKP-1/p38 MAPK信号轴,最终导致哮喘类固醇不敏感的全新机制。这一发现不仅解释了激素抵抗性哮喘的关键病理机制,更为临床治疗提供了新的潜在靶点。
背景:困扰临床的激素抵抗难题
支气管哮喘 是一种由多种炎性细胞和细胞组分参与的气道慢性炎症性疾病,全球患者人数超过3亿。
吸入性糖皮质激素是目前治疗哮喘最有效的抗炎药物之一,然而约20%的患者会出现类固醇抵抗(SR),导致对大剂量类固醇反应不佳,即类固醇不敏感(SI)以及持续的炎症,使治疗陷入困境。以往研究发现,氧化还原失衡(OS)是类固醇不敏感的重要因素,p38丝裂原活化蛋白激酶(p38 MAPK)的过度激活在这一过程中扮演核心角色。
MAPK信号通路是细胞增殖、应激、炎症、分化等信号转导通路的共同交汇通路之一。
p38 主要介导炎症、凋亡等过程,而丝裂原活化蛋白激酶磷酸酶-1 (MKP-1)则可通过去磷酸化作用使MAPK信号下调。然而,慢性氧化应激如何调控这一通路,进而导致类固醇不敏感,其上游机制尚不清楚。
项目研究
ATF3在氧化应激下的双相调节机制
研究发现,臭氧诱导的氧化应激暴露后,ATF3的调节呈现独特的时序模式。急性臭氧暴露引发快速但短暂的ATF3诱导,ATF3 mRNA表达在暴露后4小时达到峰值。然而,在长期暴露情况下,ATF3表达逐渐受到抑制。
8周慢性暴露后,ATF3蛋白和mRNA水平显著降低。同样,MKP-1表达也显著下调,而磷酸化p38水平则逐渐升高。免疫组织化学分析证实了 ATF3 的表达情况,并表明其主要存在于支气管上皮细胞中。
这种双相调节模式解释了长期处于氧化应激环境下的哮喘患者更易发展为类固醇不敏感类型的原因。
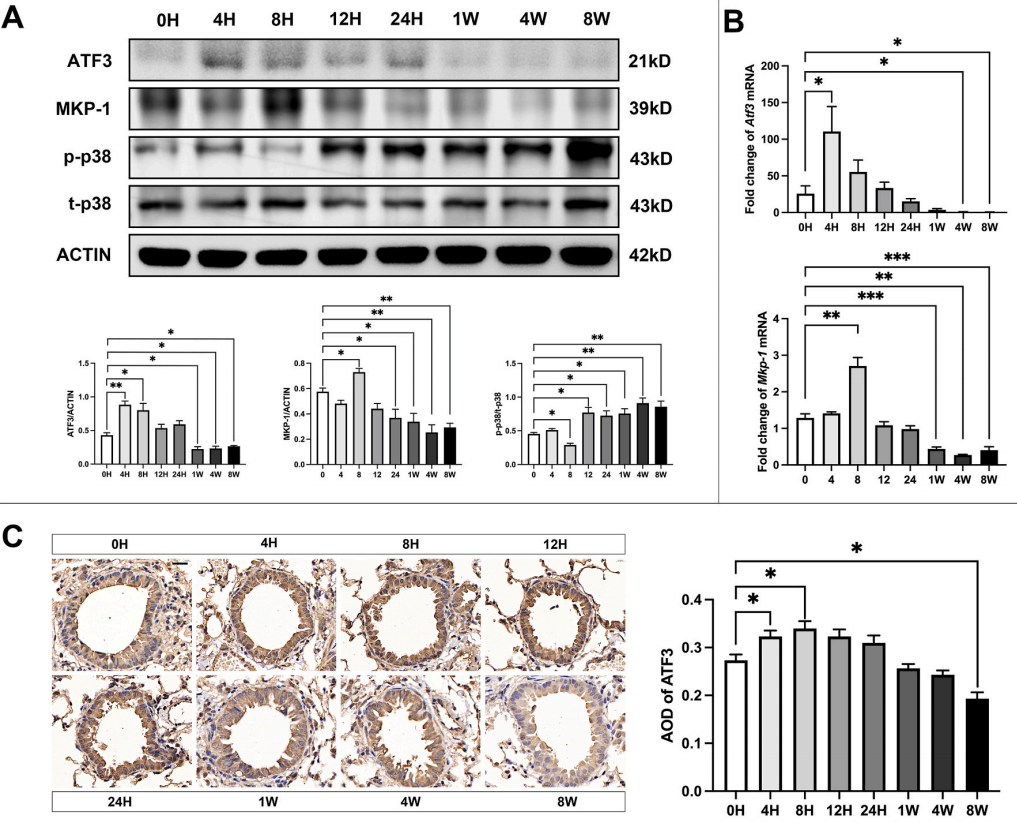
图1 慢性OS抑制小鼠肺组织中ATF3蛋白和mRNA的表达
抗氧化治疗恢复类固醇敏感性
研究显示,N-乙酰半胱氨酸(NAC)治疗可有效减轻臭氧引起的肺部氧化应激,恢复ATF3和MKP-1的表达,抑制p38 MAPK的磷酸化。此外,免疫荧光染色证实了ATF3和MKP-1蛋白在支气管上皮细胞中的共定位,这与它们之间潜在的功能相互作用一致。
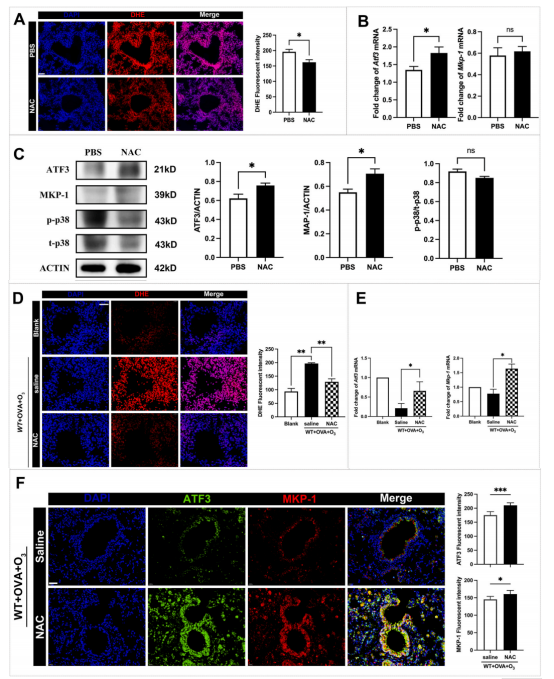
图2 在急性氧化应激实验和 SI 哮喘模型中
抑制氧化应激均增加了肺组织中 ATF3 和 MKP-1 的表达
关键的是,NAC治疗使原本对类固醇不敏感的小鼠恢复了对地塞米松(DEX)的反应性。经NAC预处理的小鼠,在接受地塞米松治疗后,小气道功能显著改善,气道炎症明显减轻。
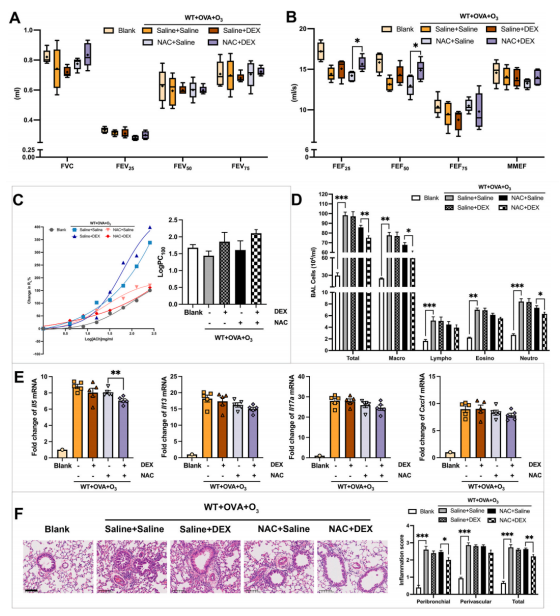
图3 在SI哮喘模型中
抑制OS可部分恢复DEX对臭氧损伤的抑制作用
ATF3基因的增强表达通过诱导 MKP-1 和抑制 p38 MAPK来恢复对类固醇的敏感性
研究人员通过AAV载体递送ATF3基因,实现了在肺组织中ATF3的增强表达。
结果显示,ATF3的过表达不仅上调了MKP-1的表达,还显著抑制了p38 MAPK的活化。免疫荧光染色也证实了这些分子变化。
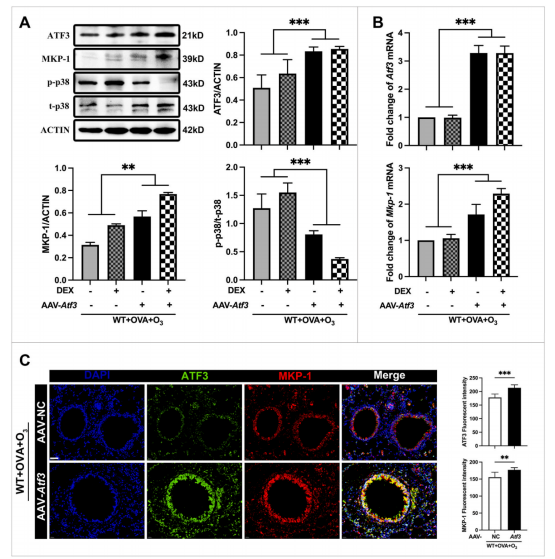
图4 增强ATF3表达可增加SI哮喘模型小鼠肺组织中
ATF3和MKP-1的表达
此外,在ATF3基因表达增强的基础上施加地塞米松治疗,可显著改善肺功能参数,减轻气道高反应性,并抑制关键炎症细胞因子的表达。这表明增强ATF3表达可能是逆转激素抵抗的有效策略。

图5 增强ATF3表达部分恢复了SI哮喘模型的类固醇敏感性
ATF3缺失通过MKP-1/ p38 MAPK信号通路降低类固醇反应性
研究进一步使用Atf3-/-敲除小鼠进行研究,确定了Atf3缺乏导致SI的机制。Western blot、qPCR和免疫荧光分析一致表明,ATF3缺失导致MKP-1表达减少和p38 MAPK磷酸化增强。相反,ATF3的基因互补有效地恢复了MKP-1的表达和减弱了p38 MAPK的磷酸化,进而证实了ATF3/ MKP-1/p38轴在维持类固醇反应性中的重要作用。
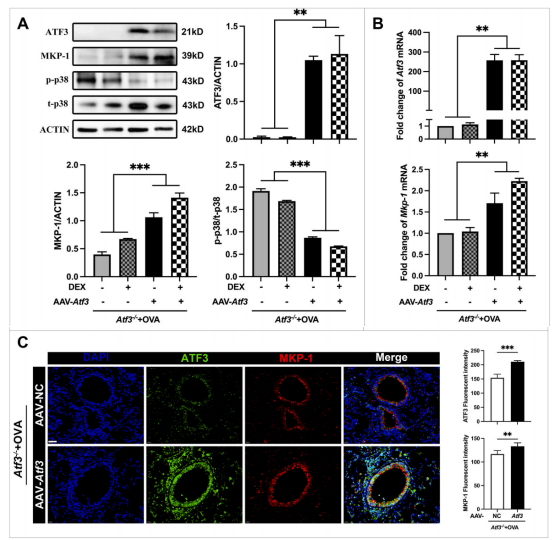
图6 ATF3的双向调控导致MKP-1的同向表达
在Atf3 - / -哮喘模型中,DEX单药治疗显示出有限的治疗效果,仅对小气道功能产生边缘改善。相反,先进行ATF3的重组再进行DEX治疗,显著增强了类固醇的疗效,从而实现了全面的肺部改善,肺功能在多个参数上都有显著提升。此外,ATF3 基因增强使 DEX 显示出广泛的抗炎作用。

图7 ATF3 的缺失会降低哮喘模型中的类固醇敏感性
ATF3-MKP-1 轴的协同作用对于 DEX 诱导的抗炎效应至关重要
在体外实验中,用 H2O2刺激 9 小时显著上调了 ATF3和MKP-1的表达,并抑制p38 MAPK的磷酸化。使用抗氧化剂NAC预处理后,尽管并未促使 ATF3表达增加,但它仍提高了 MKP-1 的表达水平并降低p38 MAPK的磷酸化水平。这提示氧化应激还可通过不依赖ATF3的途径调控MKP-1/p38通路。
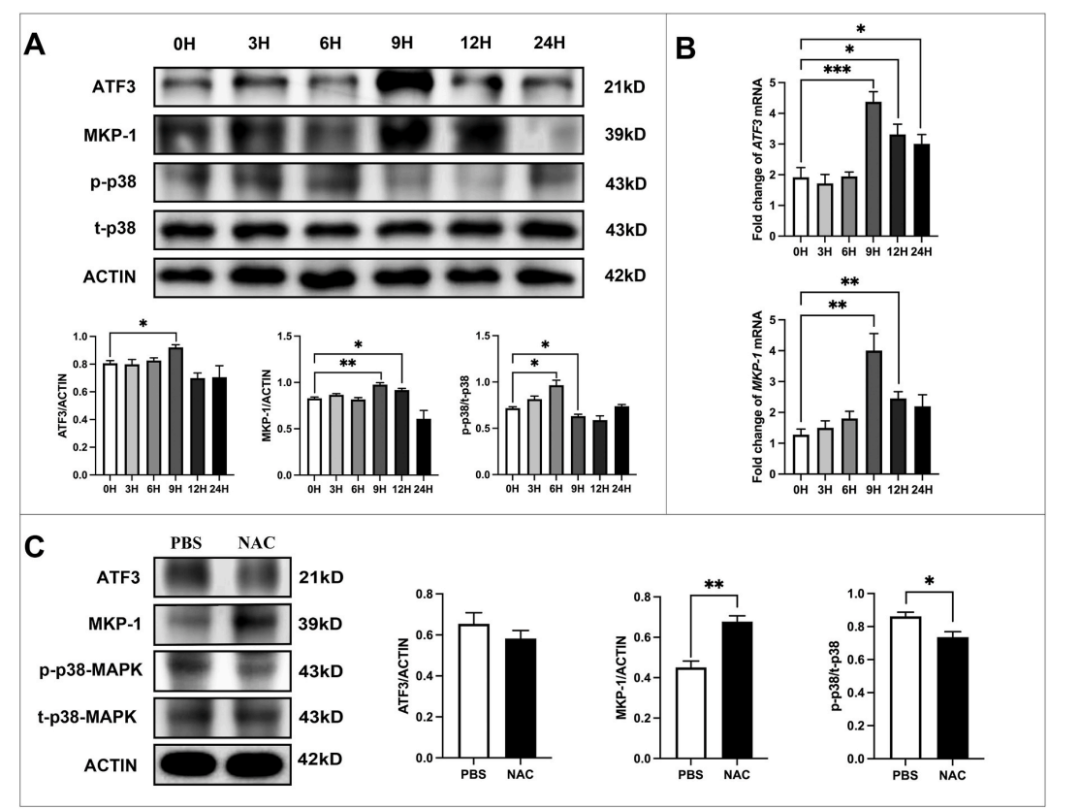
图8 H2O2通过氧化应激诱导ATF3和MKP-1表达,
并抑制p38 MAPK磷酸化
在BEAS-2B细胞模型中,研究发现ATF3和MKP-1之间存在正反馈回路。
一方面,过表达ATF3可显著增强MKP-1启动子活性,进而上调MKP-1表达;
另一方面,敲低MKP-1则会降低ATF3的表达水平。
这表明两者之间存在相互促进的调控关系,这种机制在维持氧化应激条件下的信号平衡中可能发挥着重要作用。
蛋白质印迹分析表明,ATF3和MKP-1均独立抑制p38 MAPK磷酸化,其中MKP-1是ATF3介导的p38抑制的关键下游效应因子。
为探究这一通路在激素治疗中的作用,研究人员考察了ATF3-MKP-1-p38轴在地塞米松(DEX)抑制IL-13诱导的炎症反应中的功能。
结果表明,ATF3过表达能够显著抑制TNF-α等促炎细胞因子的产生,而MKP-1敲低则会产生相反效果。尤为关键的是,当MKP-1被敲低时,ATF3过表达和DEX治疗的抗炎作用都被显著削弱,这充分说明ATF3和MKP-1在调节类固醇抗炎效果中具有功能上的相互依赖性,二者协同作用共同维持氧化应激条件下的信号平衡。
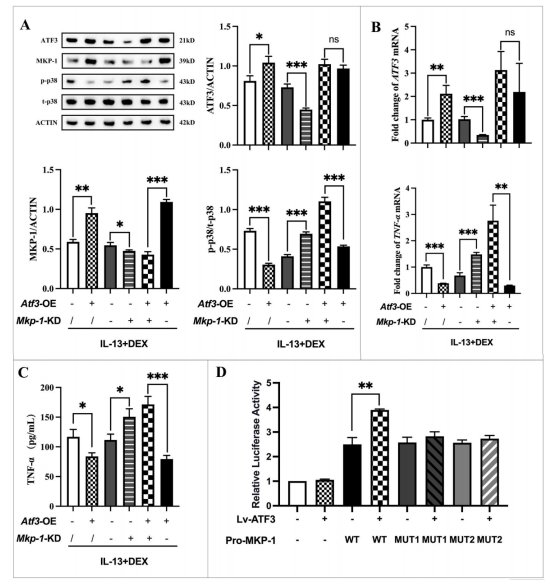
图9 ATF3对类固醇在体外抗炎作用的影响
与 MKP-1的表达有关
研究结论
该研究确定了氧化还原敏感的转录调节因子ATF3,是连接氧化应激和糖皮质激素不敏感的关键介质。
在机制上,研究发现ATF3-MKP-1-p38 MAPK轴是反应性的中心:慢性氧化应激 → ATF3表达下调 → MKP-1转录减少 → p38 MAPK磷酸化增强,进而导致哮喘中的类固醇不敏感性。这为理解激素抵抗性哮喘的发病机制提供了新视角,也为开发针对难治性哮喘的新疗法提供了重要的理论靶点。
未来研究需要进一步验证ATF3在人类哮喘患者中的表达模式及其与类固醇敏感性的关联,并探索靶向ATF3的治疗方法在临床实践中的可行性和安全性。同时,探索ATF3与其他已知激素抵抗机制(如GR异常、HDAC2活性降低等)的相互作用,也将有助于更全面地理解激素抵抗性哮喘的复杂病理网络。
吉满助力
本研究中所用的过表达慢病毒、敲低慢病毒、AAV5 载体、pGL3质粒、转染试剂和双荧光素酶报告检测试剂盒均由吉满生物(Genomeditech)提供。
了解更多质粒构建,病毒包装,稳转株构建,敲除细胞株,报告基因检测等相关服务。欢迎访问吉满生物官网:www.genomeditech.com
免费咨询电话:400-627-9288
原文引用
“AAV5 vectors carrying murine ATF3 (NM_007498.3) were constructed with homology-based seamless cloning technology under the help of Genomeditech (Shanghai) Co., Ltd.”
"To define the role of the ATF3-MKP-1 axis in the cellular response to DEX, a stable ATF3-overexpressing cell line was generated by lentiviral transduction (Lv-ATF3), and MKP-1 was knocked down using siMKP-1(Genomeditech, Shanghai, China). "
To generate a lentivirus overexpressing human ATF3 (GenBank Accession No. NM_001674.4), the full-length coding sequence of ATF3 was cloned into the PGMLV-CMV-3 × Flag-PGK-Puro lentiviral vector (Genomeditech, Shanghai, China).
The recombinant plasmid was co-transfected with the lentiviral packaging system into HEK293T cells using HG transgene reagent (Genomeditech).
At approximately 70 % confluence, cells were co-transfected with 250 ng of a promoter-reporter plasmid, 250 ng of an ATF3 expression plasmid or its empty vector control, and 25 ng of the pRL-TK plasmid (as an internal control), using 1.5 μL of GMTrans Liposomal Transfection Reagent (Genomeditech) per well according to the manufacturer's instructions.
After 48 h, cells were lysed, and luciferase activities were measured using a dual-luciferase reporter assay kit (Genomeditech).
The MKP-1 promoters (WT and mutant) were cloned into pGL3 vectors by Genomeditech, and the sequences are provided in the supplementary materials.
文献原文
https://linkinghub.elsevier.com/retrieve/pii/S0891-5849(25)01346-2







